


ISO13485 医療機器マネジメントシステム
I.広がるISO
医薬品医療機器等法との関わり
- 医薬品医療機器等法(平成26年11月25日施行)により、医療機器の製造や販売を取り巻く法規制が変わりました。
医療機器の元売である製造販売業が製造、販売、市販後を管理することは従来と同じですが、製造業に対する要件であったQMSは、改正法により製造販売業に対する要件となり、製造業は登録制になりました。
QMS適合性調査は、製造販売業に対する承認品目の製品群で行われ、その際に登録製造業者も調査を受けることになりました。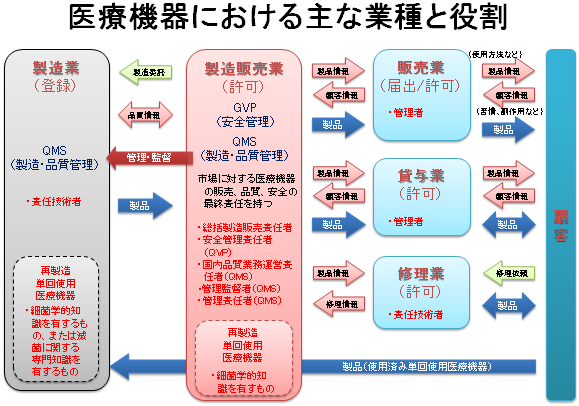
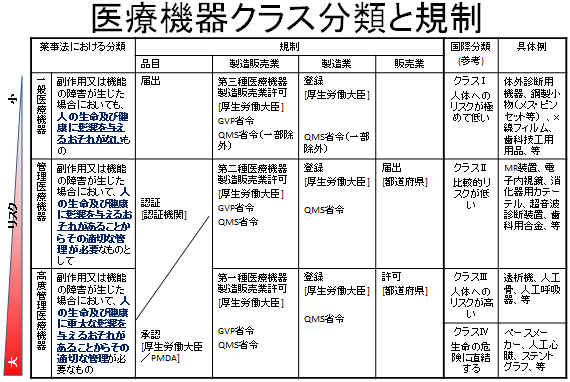
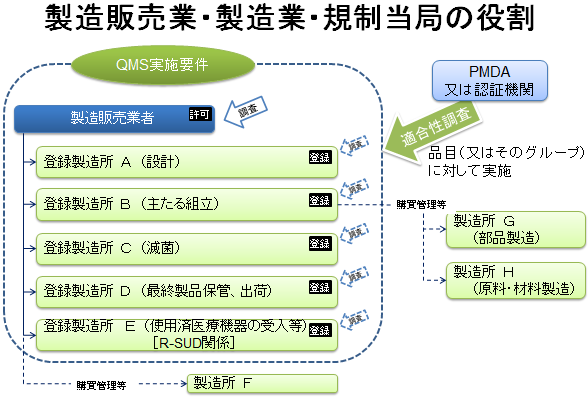
医療機器のQMSは、医療機器のグローバル化により、ISO13485の規格に準拠して策定されました。さらに、クラスⅡ(及びクラスⅢの一部)の、安全管理が軽度の医療機器については、品目承認・認証の審査を第三者機関(厚労省が認定する)が行うことになりました。その際には「QMS基準による製造及び品質管理の確認」も審査項目の一つになりますので、医療機器製造メーカーがISO13485の認証を取得しておくことのメリットは非常に大きくなりました。
ISO13485は医療機器の品質を管理する仕組みの規格 ≠ 製品の規格 - ISO13485はISO9001をベースに作られた医療機器製造、品質管理のための規格であり、製品・サービスの品質の良否を決める規格ではない。顧客が求める機能を持ち、安全性の高い一定の品質の製品・サービスが常に提供されるような仕組みを作る規格である。
既にISO9001は商取引のグローバル化を背景に、工業生産を行う全世界に浸透しているが、医療機器についても製造・販売のグローバル化を背景に、世界共通の管理システムとして日本においても、医療機器製造のGMPの仕組みとして採用され、その後、厚労省の省令によるQMS基準の仕組みに変更されました。この規格を取り入れることにより、顧客が要求する一定の品質の製品・サービスを提供するための製造管理、品質管理の仕組みが実現する。
Ⅱ.ISO13485の初歩
- ISO13485とは?
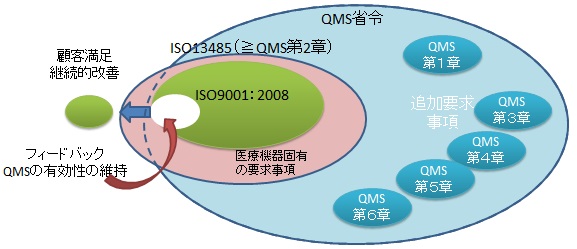
ISO9001とISO13485の要求事項の関係概略図
- この規格の目的は?
- (a) 企業が、医療機器の品質と安全性を安定的に確保するためのシステムを提供する。
- (b) 公的機関(第三者機関)が企業の医療機器製造管理及び品質管理能力を評価する場合の基準となる。(QMS要件)
- この規格の認証取得、審査機関は?
- (a) 国際標準化機構(ISO)-日本では相互認証している(財)日本適合性認定協会(JAB)より認定を受けた機関が、企業のこの規格の構築、運用状況を審査し合格すれば認証を与える。ただし、この認証取得は、JABが独自に行うもので、医療機器の品目承認・認証(厚労省管轄)には効果がない。
- (b) 厚労省が医療機器の承認・認証の第三機関として認定した機関でかつJABより認定を受けた機関が、企業のこの規格の構築、運用状況を審査し合格すれば認証を与える。この認証は、第三者機関が行う医療機器の承認・認証の際に有効である。
- この規格の内容は(実際にすることは)?
要求事項(しなければならないこと)をもれなく実行することにより、規格の目的を達成するための品質マネジメントシステムが確立するようになっている。- (a) 経営トップは品質に対する責任を明確にして、品質方針を出し、全従業員は関連する品質目標の達成に自覚を持って努力しなければならない。
- (b) この規格に従って、企業固有のやり方をマニュアル・手順書に文書化し、それに従った作業を行い、対外的な証拠となるように記録を取ること。
- (c) 運用状況をチェックし、安定的に安全な製品を市場に出せるよう、品質マネジメントシステムを改善すること。
III.ISO13485の内容
標準化→文書化→実行→記録
- やり方を決め、実行し、記録する
ISO13485は要求事項の中で、貴社がISO13485で仕事をするなら「○○についてどのようにするか決めて実施してください」と言っている。ISOでは製品を作るための仕組みを、自分たちのやり方で決めなければならない(ただしISO13485の要求事項に従って)<標準化>。やり方を決めたら、統一できるように、それを紙に書いて参照できるようにすることが重要である<文書化>(ISO13485では最低限文書化するものが決めてある)。「□□する」と言うだけで従業員に徹底するのは困難である。Aさんに聞くと◇◇するといい、Bさんは▽▽するといっている。これでは品質管理はできない。
次に決めたことを実行し<実行>、その結果を証拠として記録しなければならない<記録>(ISO13485では最低限の必要な記録が決めてある)。
「当社では○○について△△しており、それはここに決めてあります。これがその通り行った結果の記録です。そうして作られる製品・サービスがこれです。」と具体的に示すことで、真にお客様の信頼を得ることができる。
計画(標準化)Plan → 実行Do → チェックCheck → 処置・改善Act
- やり方を定め、その通り実行し、出来ているかチェックし、改善する
ISO13485の要求事項をふまえて、業務のやり方、目標、基準を決め(計画Plan)、その通り実施Doし、決められた通り、計画した通りできたかどうか、安全性を含めて顧客の要求を満たしえたかどうかチェックCheckし、できていないところ、不足があれば適切に処置し、改善Actする(PDCAサイクル)。これによって、企業の品質管理の仕組みがますますよくなる。 - ISO13485のポイント
- ISO13485の要求事項
| 条項番号 | 要求事項 | |
| 4.品質マネジメントシステム | 4.1 | 一般要求事項 |
| 4.2 | 文書化に関する要求事項 4.2.1 一般 4.2.2 品質マニュアル 4.2.3 医療機器ファイル 4.2.4 文書管理 4.2.5 記録の管理 | |
| 5.経営者の責任 | 5.1 | 経営者のコミットメント |
| 5.2 | 顧客重視 | |
| 5.3 | 品質方針 | |
| 5.4 | 計画 5.4.1 品質目標 5.4.2 品質マネジメントシステムの計画 | |
| 5.5 | 責任、権限及びコミュニケーション 5.5.1 責任及び権限 5.5.2 管理責任者 5.5.3 内部コミュニケーション | |
| 5.6 | マネジメントレビュー 5.6.1 一般 5.6.2 マネジメントレビューへのインプット 5.6.3 マネジメントレビューからのアウトプット | |
| 6.資源の運用管理 | 6.1 | 資源の提供 |
| 6.2 | 人的資源 6.2.1 一般 6.2.2 力量、認識及び教育・訓練 | |
| 6.3 | インフラストラクチャ | |
| 6.4 | 作業環境及び汚染の管理 6.4.1 作業環境 6.4.2 汚染の管理 | |
| 7.製品実現 | 7.1 | 製品実現の計画 |
| 7.2 | 顧客関連のプロセス 7.2.1 製品に関連する要求事項の明確化 7.2.2 製品に関連する要求事項のレビュー 7.2.3 顧客とのコミュニケーション | |
| 7.3 | 設計・開発 7.3.1 一般 7.3.2 設計・開発の計画 7.3.3 設計・開発へのインプット 7.3.4 設計・開発からのアウトプット 7.3.5 設計・開発のレビュー 7.3.6 設計・開発の検証 7.3.7 設計・開発のバリデーション 7.3.8 設計・開発の移管 7.3.9 設計・開発の変更管理 7.3.10 設計・開発のファイル | |
| 7.4 | 購買 7.4.1 購買プロセス 7.4.2 購買情報 7.4.3 購買製品の検証 | |
| 7.5 | 製造及びサービス提供 7.5.1 製造及びサービス提供の管理 7.5.2 製品の清浄性 7.5.3 据付け活動 7.5.4 付帯サービス活動 7.5.5 滅菌医療機器に対する特別要求事項 7.5.6 製造及びサービス提供に関するプロセスバリデーション 7.5.7 滅菌及び無菌バリアシステムのプロセスバリデーションに対する特別要求事項 7.5.8 識別 7.5.9 トレーサビリティ 7.5.9.1 一般 7.5.9.2 埋込み医療機器に対する特別要求事項 7.5.10 顧客の所有物 7.5.11 製品の保存 | |
| 7.6 | 監視機器及び測定機器の管理 | |
| 8.測定、分析及び改善 | 8.1 | 一般 |
| 8.2 | 監視及び測定 8.2.1 フィードバック 8.2.2 苦情処理 8.2.3 規制当局への報告 8.2.4 内部監査 8.2.5 プロセスの監視及び測定 8.2.6 製品の監視及び測定 | |
| 8.3 | 不適合品の管理 8.3.1 一般 8.3.2 引き渡し前の不適合製品における処置 8.3.3 引き渡し後に発見された不適合製品における処置 8.3.4 手直し | |
| 8.4 | データの分析 | |
| 8.5 | 改善 8.5.1 一般 8.5.2 是正処置 8.5.3 予防処置 | |
ISO13485の仕組みを作り上げるにはどうすればよいか
- 基本的には企業のありのままを決まりとする
ISOを導入すると記録が増えて仕事が煩雑になる、だから……とよく言われる。ISO13485の要求事項の中にはこれまでやってこなかった項目があるかもしれない。確かにそれについては、新たにやり方を決めて、実行し、記録しなければならないから仕事が増える。
しかし、それぞれの企業にはこれまで信用ある製品・サービスを顧客に提供してきた実績がある。現在、企業でやっている通り自信を持って決まりにすることが重要である。不足しているところがあれば加え、変更が必要になれば必要なところを変えるようにすることが肝要である。やれないことを決めないことも重要。
IV.導入のスケジュール
まず何よりも経営トップの不退転の決意とリーダーシップが必要である。忙しさを理由に後回しや、中断すると再開は大変難しい。タイミングをはかり、やるとなったらやり遂げる覚悟を経営者自身が持たなければならない。
一般化すると次図のように、ほぼ12ヶ月の活動になるが、期間は各社それぞれの規模や業務内容、取得活動の程度により異なる。
ISO13485構築基本プログラム

認証取得活動標準スケジュール
| 支援項目/活動項目 | 経過月 | |||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| ISO13485規格要求事項の理解 | ■ | |||||||||||
| 現状把握・現工程の標準化 | ■ | ■ | ■ | |||||||||
| 目標設定、実行計画の策定 | ||||||||||||
| 文書作成(システム構築) | ■ | ■ | ||||||||||
| ・品質マニュアルの作成 | ■ | ■ | ||||||||||
| ・規定類の作成 | ■ | ■ | ■ | |||||||||
| ・手順書類、帳票の整備、作成 | ■ | ■ | ■ | |||||||||
| システムの運用 | ★ | ★ | ★ | ★ | ★ | ★ | ||||||
| システムの教育(運用状況チェック) | ■ | ■ | ■ | ■ | ■ | ■ | ||||||
| ・内部監査員養成(2日間) | ■ | |||||||||||
| ・内部監査実施 | ★ | |||||||||||
| ・マネジメントレビュー | ★ | |||||||||||
| 審査対策 | ■ | ■ | ||||||||||
| 審査後のフォロー | ■ | |||||||||||
| ■:コンサルタントが支援 ★:貴社で実施 ●:審査対応 | ||||||||||||
| 申請 | ● | |||||||||||
| 登録1次審査 | ● | |||||||||||
| 登録2次審査 | ● | |||||||||||
| 認証取得 | ● | |||||||||||